

Mukopolisacharydoza to jedna z około 7 tysięcy chorób rzadkich. Na świecie zarejestrowanych jest 7 typów tej choroby. Nazwa choroby pochodzi od mukopolisacharydów - łańcuchów cząstek cukru biorących udział w tworzeniu tkanki łącznej. Za pomocą odpowiednich enzymów są one w organizmie nieustannie rozkładane i ponownie tworzone. U osób chorych na mukopolisacharydozę ze względu na brak odpowiedniego enzymu proces ten jest zaburzony.
- Mój syn ma rzadką chorobę, której nazwy często nie znają nawet lekarze, bo w Polsce ma ją zaledwie kilkanaścioro dzieci: mukopolisacharydoza typ 1, w skrócie MPS. Błąd genu powoduje, że w organizmie brakuje enzymu rozkładającego mukopolisacharydy. Odkładają się one we wszystkich narządach ciała, powoli je niszcząc – opowiada Magda, mama chłopca chorego na MPS.
Mukopolisacharydoza jest chorobą postępującą o różnorodnym przebiegu, szerokim spectrum objawów i różnym ich nasileniu. Wiedza na temat choroby, ma kluczowe znaczenie dla poprawy sytuacji pacjentów, ponieważ ciągle dużym problemem pozostaje właściwa diagnostyka.
- Choć MPS jest chorobą genetyczną, jej objawy pojawiają się z czasem. Początkowo dzieci rozwijają się tak jak ich zdrowi rówieśnicy. Najczęściej wstępne diagnozy stawiane są między 18 a 36 miesiącem życia, kiedy w komórkach dochodzi do nadmiernego gromadzenia się sacharydów. Uwagę mogą zwrócić dorosły wygląd twarzy: ciemne brwi, szeroko rozstawione oczy oraz powiększona wątroba – mówi Prof. nadzw. dr hab. n. med. Zbigniew Żuber, kierownik Oddziału Dzieci Starszych z Pododdziałem Neurologicznym, Reumatologicznym, Rehabilitacyjnym Szpitala Dziecięcego św. Ludwika w Krakowie.
W Polsce potwierdzono występowanie V typów Mukopolisacharydozy. Każdy typ charakteryzuje się brakiem innego enzymu.
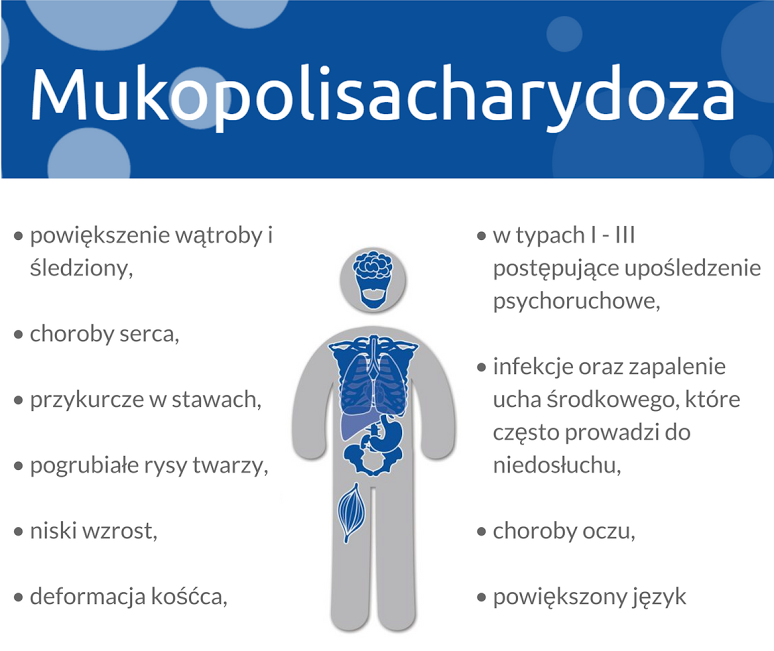
Jak podkreśla profesor Żuber, cechami wspólnymi dla wszystkich typów MPS są:
powiększenie wątroby i śledziony,
choroby serca,
przykurcze w stawach,
pogrubiałe rysy twarzy,
niski wzrost,
deformacja kośćca,
w typach I - III postępujące upośledzenie psychoruchowe,
infekcje oraz zapalenie ucha środkowego, które często prowadzi do niedosłuchu,
choroby oczu,
powiększony język
Aby sprawdzić jak wygląda diagnostyka typu I mukopolisacharydozy – jednego z najwcześniej opisanych typów choroby - Royal Manchester Children's Hospital we współpracy z Sanofi Genzyme i organizacją Society for Mucopolysaccharide Diseases, przeprowadziły międzynarodowe badanie ankietowe wśród lekarzy i opiekunów pacjentów. Z badania wynika, że od momentu wystąpienia pierwszych objawów, do postawienia diagnozy mijają średnio 2 lata i 7 miesięcy. W tym czasie pacjenci odwiedzają średnio 5 lekarzy. Mukopolisacharydoza typu I najczęściej mylona przez lekarzy z reumatoidalnym zapaleniem stawów, a także z innymi chorobami takimi jak np. toczeń.
Więcej informacji na temat MPS odnaleźć można na stronie internetowej www.chorobyspichrzeniowe.pl
Poznajcie też historię Michała




















